Summary statistics on BENGI GM12878 benchmarks¶
Context¶
We computed summary statistics on the BENGI benchmarks in order to ensure that we find the same as the authors.
Remarks¶
The R code we used to compute the results below is detailed here.
Introduction¶
$ head GM12878.CHiC-Benchmark.v4.txt EH37E0466501 ENSG00000206181.4 1 cv-2 EH37E0466501 ENSG00000267587.1 0 cv-2 ... # <enhancer_id> <gene_id (hg19)> <interaction?> <cross_val_group>
First we verify that the reported genes ids are not degenerate with respect to reported gene version.
$ awk '!id[$2]++ {print $2}' GM12878.CHiC-Benchmark.v4.txt |wc -l 28455 -> number of unique gene ids (with gene version)$ awk 'BEGIN{FS="\t"} {split($2,id,"."); if(!complete_id[$2]){nb_versions[id[1]]++}; complete_id[$2]++} END{for(u in nb_versions){print u, nb_versions[u]}}' GM12878.CHiC-Benchmark.v4.txt |wc -l 28455 -> number of unique gene ids (without gene versions)
=> seems ok, only one version / gene at least in that particular benchmark. Indeed,
awk 'BEGIN{FS="\t"} {split($2,id,"."); if(!complete_id[$2]){nb_versions[id[1]]++}; complete_id[$2]++} END{for(u in nb_versions){if(nb_versions[u]>1){print u, nb_versions[u]}}}' GM12878.CHiC-Benchmark.v4.txt
yields nothing.
Overlap coefficient¶
Remark: there are more (up to 4 times more) positives with natural ratio than with fixed ratio, as explained p. 13 in the paper § Generation of Negative pairs.
"For the fixed ratio datasets, we also excluded genes that were in the positi ve pairs for the cCREs-ELS in other BENGI datasets before randomly selecting the negatives. If a cCRE-ELS exhibited fewer than four negative pairs, then it was excluded from this fixed ratio set."
Excluding positives from the other BENGI datasets before selecting the negatives possibly leads to less than 4 negatives remaining for each positive, meaning some positives must be excluded (later on we shall check this).
Natural ratio


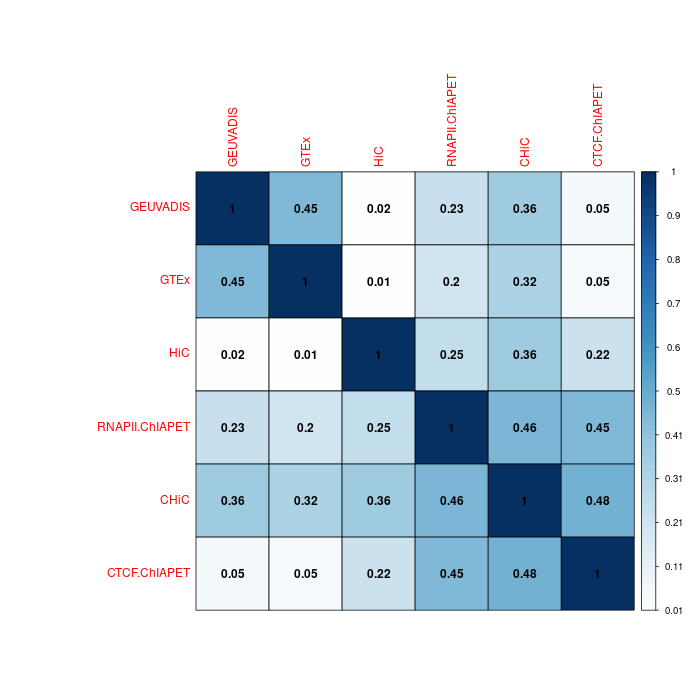
Fixed ratio

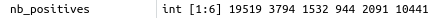
Version A: all pairs for 3D chromatin interactions, natural ratio¶
Same as (Fig. 2a) in the paper => OK.
Version B: all pairs for 3D chromatin interactions, fixed ratio¶
Clearly less overlapping between datasets with fixed ratio.
Distance distribution¶
Summary statistics on raw BENGI datasets¶
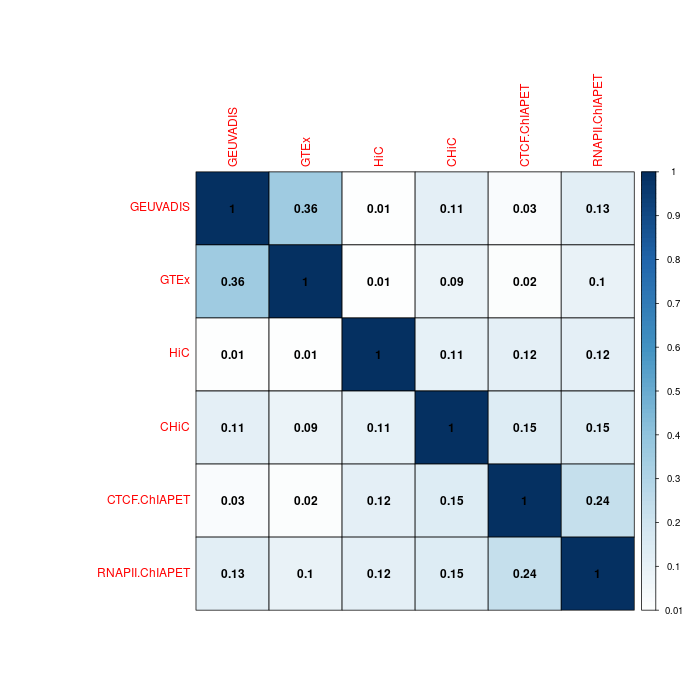
0.95-quantiles of distance distribution of the 6 GM12878 benchmarks
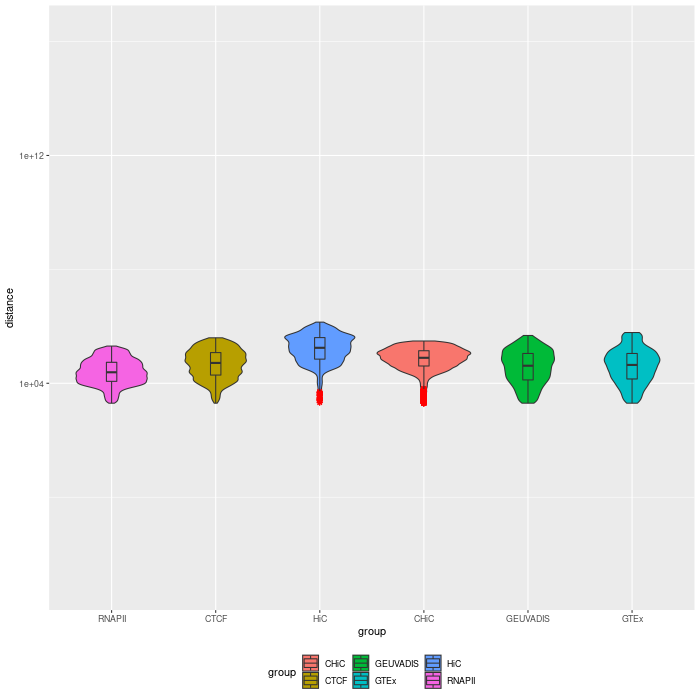
Distance distribution of enhancer-gene interactions
In order to have more reliable results and to facilitate further analysis, we reprocessed data to have all useful information in a unique .bedpe file per benchmark. We use a Python script to that purpose, BENGI_to_bedpe.py (see dedicated notebook/guidelines, for instance: BENGI_to_custom_bedpe.
Distance distributions using authors calculate.distance.py¶
Python2 required.
conda activate py2
Warning: it is not possible to use the script directly without modifying it as the enhancer dictionary is built by default with rDHS accession instead of ccRE accession ([3] instead of [4]). So we first have to replace line 20:
enhancerDict[line[3]]=[int(line[1]),int(line[2])]
with the following:
enhancerDict[line[4]]=[int(line[1]),int(line[2])]
Now:
python local_Scripts/Generate-Benchmark/calculate.distance.py ~/Documents/INSERM/BENGI/Benchmark/Annotations/GENCODEv19-TSSs.bed ~/Documents/INSERM/BENGI/Benchmark/Annotations/hg19-cCREs.bed ~/Documents/INSERM/BENGI/Benchmark/All-Pairs.Natural-Ratio/GM12878.CHiC-Benchmark.v3.txt ~/Documents/INSERM/BENGI/Benchmark/All-Pairs.Natural-Ratio/GM12878.CHiC-Benchmark.v3.distances.txt
It worked correctly and yielded:
289224.0 # (print np.percentile(distanceArray,95))
CHiC: 289224.0
CTCF-ChIAPET: 373167.75
GEUVADIS: 454695.2
GTEx: 570084.45
HiC: 1322447.9000000001
RNAPII-ChIAPET: 191565.0