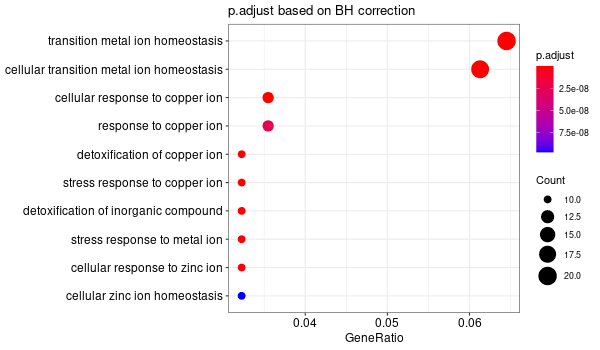
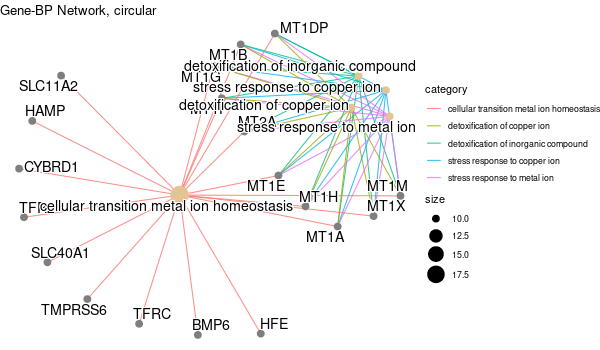
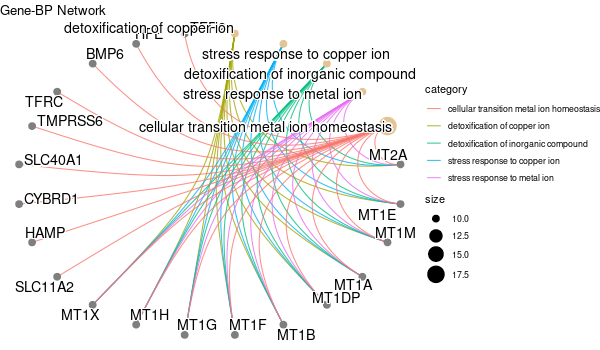
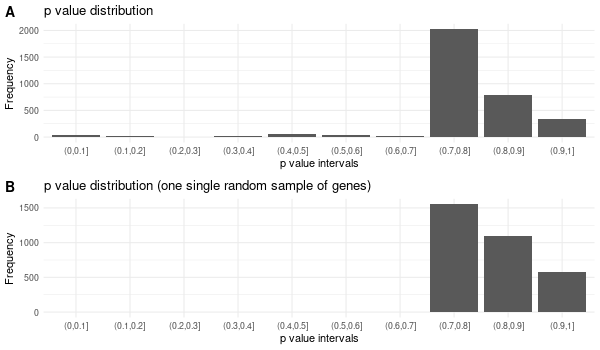
New custom script to compute GO enrichment: GO_enrichment_FDR.R¶
We adapt GO_enrichment.R so that the multiple testing correction is less stringent: we want to control the FDR, rather than the FWER with the Šidák procedure.
We name our script GO_enrichment_FDR.R.
We should look at the enrichGO function from the clusterProfiler library:
https://rdrr.io/bioc/clusterProfiler/man/enrichGO.html
https://www.rdocumentation.org/packages/clusterProfiler/versions/3.0.4/topics/enrichGO
https://yulab-smu.top/biomedical-knowledge-mining-book/clusterprofiler-go.html
Dependencies¶
We use R 4.0. We need the clusterProfiler package.
The following does not work, so all this quotation is just to keep a record in case we encounter similar problems:
BiocManager::install("clusterProfiler")Installation paths not writeable, unable to update packages path: /usr/lib/R/library packages: boot, class, cluster, foreign, KernSmooth, lattice, MASS, Matrix, mgcv, nlme, nnet, spatial, survival.libPaths()[1] "/home/thoellinger/R/x86_64-pc-linux-gnu-library/4.0" [2] "/usr/local/lib/R/site-library" [3] "/usr/lib/R/site-library" [4] "/usr/lib/R/library"BiocManager::install(c("boot", "class", "cluster", "foreign", "KernSmooth", "lattice", "MASS", "Matrix", "mgcv", "nlme", "nnet", "spatial", "survival"), lib = "/home/thoellinger/R/x86_64-pc-linux-gnu-library/4.0")BiocManager::install("clusterProfiler", lib = "/home/thoellinger/R/x86_64-pc-linux-gnu-library/4.0")Error in get(name, envir = asNamespace(pkg), inherits = FALSE) : object 'get_fun_from_pkg' not found Error: unable to load R code in package ‘clusterProfiler’ Execution halted ERROR: lazy loading failed for package ‘clusterProfiler’ * removing ‘/home/thoellinger/R/x86_64-pc-linux-gnu-library/4.0/clusterProfiler’ The downloaded source packages are in ‘/tmp/Rtmp3Xmv24/downloaded_packages’ Warning message: In .inet_warning(msg) : installation of package ‘clusterProfiler’ had non-zero exit statusStill does not work. It appears that the problem is the dependency in the
rvchecklibrary, that contained theget_fun_from_pkgup to version 1.8 but does not contain it anymore ; and the authors ofclusterProfilerhave not updated their dependencies: https://www.biostars.org/p/9491480/
We need to install an older version of rvcheck, namely, the version 1.8. See details here. We download it and install it from source.
cd Documents/shared/sources
wget https://cran.microsoft.com/snapshot/2020-04-20/src/contrib/rvcheck_0.1.8.tar.gz .
install.packages("/home/thoellinger/Documents/shared/sources/rvcheck_0.1.8.tar.gz", repos = NULL, type="source")
So now:
BiocManager::install("clusterProfiler", lib = "/home/thoellinger/R/x86_64-pc-linux-gnu-library/4.0")
should work BUT one have to select "no" when the prompt asks whether to update the rvcheck package (even if it asks whether to update or not a lot of packages including rvcheck)!
Note that one might also need to install org.Hs.eg.db separately.
BiocManager::install("org.Hs.eg.db", lib = "/home/thoellinger/R/x86_64-pc-linux-gnu-library/4.0")
Again, it is necessary to select "no" when the prompt asks whether to update the rvcheck package (even if it asks whether to update or not a lot of packages including rvcheck)!
For graphics, we need a few more packages:
BiocManager::install("ggupset", lib = "/home/thoellinger/R/x86_64-pc-linux-gnu-library/4.0")
BiocManager::install("ggnewscale", lib = "/home/thoellinger/R/x86_64-pc-linux-gnu-library/4.0")
Again, it is necessary to select "no" when the prompt asks whether to update the rvcheck package.
We followed this guide for visual outputs: https://yulab-smu.top/biomedical-knowledge-mining-book/enrichplot.html
Script¶
The main part of the script will use the groupGO function that basically does everything:
res = groupGO(gene = U, # vector of entrez gene id
OrgDb = org.Hs.eg.db, # human organism
ont = "BP", # ontology, "BP", "MF" or "CC"
universe = universe, # background genes (entrez)
pvalueCutoff = 0.05,
pAdjustMethod = "BH", # one of "holm", "hochberg", "hommel", "bonferroni",
# "BH", "BY", "fdr", "none"
readable = TRUE) # whether mapping gene ID to gene Name
The OrgDb parameter is an object specifying the organism of interest. For human, one must use OrgDb = org.Hs.eg.db. Note that OrgDb object are available in Bioconductor for 20 organisms: http://bioconductor.org/packages/release/BiocViews.html#___OrgDb
One can find the list of types of sequence id available (for instance "ENTREZID", "ENSEMBL", "REFSEQ") using columns(org.Hs.eg.db):
> columns(org.Hs.eg.db) [1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT" "ENSEMBLTRANS" "ENTREZID" "ENZYME" "EVIDENCE" "EVIDENCEALL" "GENENAME" "GO" "GOALL" [13] "IPI" "MAP" "OMIM" "ONTOLOGY" "ONTOLOGYALL" "PATH" "PFAM" "PMID" "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG" [25] "UNIGENE" "UNIPROT"
Here is the full content of GO_enrichment_FDR.R:
################## # OPTION PARSING # ################## suppressPackageStartupMessages(library("optparse")) option_list <- list( make_option(c("-k", "--key"), default="ENTREZID", help="type of gene identifiers used in other arguments (one of \"ENTREZID\", \"ENSEMBL\", \"REFSEQ\", \"GENENAME\", \"SYMBOL\") [default=%default]"), make_option(c("-u", "--universe"), default="None", help="list of background gene identifiers (of same type as --key), WITHOUT header. Use \"None\" for default background (given by the clusterProfiler package) [default=%default]"), make_option(c("-G", "--genes"), default="stdin", help="list of foreground gene identifiers (of same type as --key), WITHOUT header [default=%default]"), make_option(c("-c", "--ontology"), help="choose the GO category < BP | MF | CC > [default=%default]", default="BP"), make_option(c("-p", "--pval"), help="p-value threshold. All GO terms found to be enriched up to this threshold will be saved as output. [default=%default]", default=0.1), make_option(c("-q", "--qval"), help="q-value threshold. All GO terms found to be enriched up to this threshold will be saved as output. [default=%default]", default=0.2), make_option(c("-a", "--padjMethod"), help="p-value adjustement method threshold. One of \"holm\", \"hochberg\", \"hommel\", \"bonferroni\", \"BH\", \"BY\", \"fdr\", \"none\" [default=%default]", default="fdr"), make_option(c("-f", "--fdr"), help="FDR (or adjusted p-value in general) threshold. All GO terms found to be enriched up to this threshold will be saved as output in a separate table. [default=%default]", default=0.1), make_option(c("-v", "--verbose"), default=TRUE, help="Verbosity (TRUE/FALSE) [default=%default]"), make_option(c("-o", "--output"), default="out", help="additional tags for otuput [default=%default]"), make_option(c("-d", "--output_dir"), default="./GO_output/", help="directory for the output [default=%default]") ) parser <- OptionParser(usage = "%prog [options] file", option_list=option_list) arguments <- parse_args(parser, positional_arguments = TRUE) opt <- arguments$options verbose = opt$verbose ################################# # Load libraries & Import data # ################################# if(verbose){ library("clusterProfiler") library("org.Hs.eg.db") library(enrichplot) library(ggplot2) } else{ suppressPackageStartupMessages(library("clusterProfiler")) suppressPackageStartupMessages(library("org.Hs.eg.db")) suppressPackageStartupMessages(library(enrichplot)) suppressPackageStartupMessages(library(ggplot2)) } if (verbose) print("Loading input data...") key = opt$key if(opt$universe=="None"){ universe = NA print("Warning: using defaut universe automatically provided by the clusterProfiler package") } else{ U = read.table(opt$universe, h=F, col.names='hs') universe = as.character(unique(U$hs)) } G = read.table(opt$genes, h=F, col.names='hs') genes = unique(G$hs) ontology = opt$ontology pval = opt$pval qval = opt$qval pAdjMethod = opt$padjMethod fdr_threshold = opt$fdr if (verbose) print("Done.") ###################### # Compute enrichment # ###################### to_readable = TRUE if (key=="SYMBOL") to_readable = FALSE if (verbose) print("Computing GO enrichment...") res = enrichGO(gene = genes, OrgDb = org.Hs.eg.db, keyType = key, ont = ontology, universe = universe, pvalueCutoff = pval, qvalueCutoff = qval, pAdjustMethod = pAdjMethod, readable = to_readable) if (verbose) print("Done.") universe_found = res@universe ################### # Compute results # ################### if(opt$universe=="None"){ sprintf("%s (default) background genes", length(res@universe)) } else{ sprintf("%s provided background genes; %s with a corresponding %s id", nrow(U), length(universe), key) } n_genes_found = strsplit(res@result$GeneRatio[[1]],'/')[[1]][2] sprintf("%s provided genes; %s found by `enrichGO`", nrow(G), n_genes_found) sprintf("Computed GO enrichment (whether significant or not) for %s distinct GO terms", nrow(res@result)) sprintf("Of those %s GO terms, %s have a %s-adjusted p-val < %s", nrow(res@result), nrow(res@result[res@result$p.adjust<=fdr_threshold,]), pAdjMethod, fdr_threshold) ################ # Write output # ################ dir.create(opt$output_dir, showWarnings = FALSE, recursive = TRUE) output = sprintf("%s/%s.%s.%s%s", opt$output_dir, opt$output, opt$ontology, opt$fdr, opt$padjMethod) if (verbose) print("Writing outputs tables...") # Full table write.table(res@result, file=sprintf("%s.tsv", output), quote=F, sep="\t", row.names=F) # Most significant GO terms write.table(res@result[res@result$p.adjust<=fdr_threshold,], file=sprintf("%s.significant.tsv", output), quote=F, sep="\t", row.names=F) if (verbose) print("Done. Writing output images...") png(file=sprintf("%s.upset.png", output), width=600, height=350) upsetplot(res) + ggtitle("UpSet Plot. Number of genes per significant GO term") graphics.off() png(file=sprintf("%s.dotplot.png", output), width=600, height=350) dotplot(res) + ggtitle(sprintf("p.adjust based on %s correction", pAdjMethod)) graphics.off() png(file=sprintf("%s.gene-concept.png", output), width=600, height=350) cnetplot(res, categorySize="pvalue", foldChange=genes, colorEdge = TRUE) + ggtitle(sprintf("Gene-%s Network, circular", ontology)) graphics.off() png(file=sprintf("%s.gene-concept.circular.png", output), width=600, height=350) cnetplot(res, categorySize="pvalue", foldChange=genes, circular = TRUE, colorEdge = TRUE) + ggtitle(sprintf("Gene-%s Network", ontology)) graphics.off() png(file=sprintf("%s.heatplot.png", output), width=600, height=350) heatplot(res) + ggtitle("Heatplot: significant GO terms against contributing genes") graphics.off() pbar <- function(x) { pi=seq(0, 1, length.out=11) mutate(x, pp = cut(p.adjust, pi)) %>% group_by(pp) %>% summarise(cnt = n()) %>% ggplot(aes(pp, cnt)) + geom_col() + theme_minimal() + xlab("p value intervals") + ylab("Frequency") } if (verbose) print("Writing last output image (this one might take some time)...") random_genes = sample(universe_found, as.integer(n_genes_found)) enrich_ref = enrichGO(gene = random_genes, OrgDb = org.Hs.eg.db, keyType = key, ont = ontology, universe = universe, pvalueCutoff = pval, qvalueCutoff = qval, pAdjustMethod = pAdjMethod, readable = to_readable) p1 <- pbar(res) + ggtitle("p value distribution") p2 <- pbar(enrich_ref) + ggtitle("p value distribution (one single random sample of genes)") png(file=sprintf("%s.pvalues.png", output), width=600, height=350) cowplot::plot_grid(p1, p2, ncol=1, labels = LETTERS[1:2]) graphics.off() if (verbose) print("Done.") q(save='no')
Example of use¶
module load system/R-4.0.4_gcc-9.3.0
> .libPaths() [1] "/home/thoellinger/R/x86_64-pc-linux-gnu-library/4.0" [2] "/tools/R/R-4.0.4_gcc-9.3.0/lib64/R/library"
install.packages("/work2/project/regenet/workspace/thoellinger/shared/sources/rvcheck_0.1.8.tar.gz", repos = NULL, type="source", lib="/home/thoellinger/R/x86_64-pc-linux-gnu-library/4.0")
BiocManager::install("clusterProfiler", lib = "/home/thoellinger/R/x86_64-pc-linux-gnu-library/4.0")
It is necessary to select "no" when the prompt asks whether to update the rvcheck package (even if it asks whether to update or not a lot of packages including rvcheck)!
We might also have to install
org.Hs.eg.dbseparately:BiocManager::install("org.Hs.eg.db", lib = "/home/thoellinger/R/x86_64-pc-linux-gnu-library/4.0")Again, it is necessary to select "no" when the prompt asks whether to update the
rvcheckpackage (even if it asks whether to update or not a lot of packages includingrvcheck)!
Finally:
mkdir -p GO_FDR/all_genes/symbol
Rscript GO_enrichment_FDR.R -k "SYMBOL" -G results/new_genes_v7.list -f 0.05 -c "BP" -a "BH" -o "default_universe" -d "GO_FDR/all_genes/symbol"
... [1] "Loading input data..." [1] "Warning: using defaut universe automatically provided by the clusterProfiler package" [1] "Done." [1] "Computing GO enrichment..." `universe` is not in character and will be ignored... [1] "Done." [1] "18866 (default) background genes" [1] "422 provided genes; 310 found by `enrichGO`" [1] "Computed GO enrichment (whether significant or not) for 3344 distinct GO terms" [1] "Of those 3344 GO terms, 29 have a BH-adjusted p-val < 0.05" [1] "Writing outputs tables..." [1] "Done. Writing output images..." ... [1] "Done."