Evaluation of Moore et al 's Distance Method over Fulco et al CRISPRi-FlowFISH validation dataset¶
Requirements¶
Python 2forrank.distance.pywhich is called byRun-Distance-Method.sh- the CRISPRi-FlowFish validation dataset from Fulco et al 2019, which is composed of 109 ground positives + 3752 ground negatives, see introduction
- list of all ccRE
- TSS annotation
Data preprocessing¶
We start with the following:
$workDir/CRISPRi_FlowFISH/
└── k562
├── 109.fulco.positives.tsvbas
├── 3754.fulco.negatives.tsv
├── 3863.fulco.tsv
├── fulco_2019_som.pdf
├── fulco_natgenet_2019_ep_reg_1000s_crispr.pdf
├── fulco_somtables.xlsx
├── README.sh
├── tableS3a.tsv
└── tableS6a.fromrow2.tsv
$ head -n 3 CRISPRi_FlowFISH/k562/3863.fulco.tsvinteraction chr start end Normalized.HiC.Contacts Activity ABC.Score 1 chr8 128969925 128972445 14.38244355 4.994596 0.00562307801 1 chr8 128973565 128976065 14.38244355 8.63099 0.009717048201
We must convert 3863.fulco.tsv in the suitable file format for Run-Distance-Method.sh, which is as follows:
$ head -n 1 Benchmark/All-Pairs.Natural-Ratio/GM12878.CHiC-Benchmark.v3.txtEH37E0279866 ENSG00000271614.1 1 cv-3
Hence we will have to bedtools intersect the validation dataset with ccRE. First we need to convert the validation dataset to bedpe, so we do:
tail -n +2 3863.fulco.tsv > 3863.fulco.noheader.tsv
awk 'BEGIN{FS="\t"; OFS="\t"} {if(NR==FNR){gnid[$2]=$1; next}; print $2, $3, $4, ".", -1, -1, $2":"$3"-"$4"::"$5, $8, ".", ".", $1, gnid[$5]}' /work2/project/fragencode/data/species.bk/homo_sapiens/hg19.gencv19/homo_sapiens.gnid.gnname.tsv 3863.fulco.noheader.tsv > 3863.fulco.bedpe
yielding:
head -n 5 3863.fulco.bedpechr8 128969925 128972445 . -1 -1 chr8:128969925-128972445::MYC 0.00562307801 . . 1 ENSG00000136997.10 chr8 128973565 128976065 . -1 -1 chr8:128973565-128976065::MYC 0.009717048201 . . 1 ENSG00000136997.10 chr8 130702125 130704625 . -1 -1 chr8:130702125-130704625::MYC 0.04272163371 . . 1 ENSG00000136997.10 chr8 130709145 130711645 . -1 -1 chr8:130709145-130711645::MYC 0.006183403862 . . 1 ENSG00000136997.10 chrX 48641372 48641493 . -1 -1 chrX:48641372-48641493::GATA1 0.0921316669 . . 1 ENSG00000102145.9
Problem: for 3 lines, no gene id is found, as we see with:
awk 'BEGIN{FS="\t"} {occurence[$12]++} END{for(u in occurence){print u, occurence[u]}}' 3863.fulco.bedpe |sort -rnk2,2
Moreover, 2 of these 3 lines are ground positives:
awk 'BEGIN{FS="\t"} {if($12==""){print $0}}' 3863.fulco.bedpechr8 130701606 130701940 . -1 -1 chr8:130701606-130701940::PVT1-TSS1 0.002177529321 . . 1
chr8 130704704 130705463 . -1 -1 chr8:130704704-130705463::PVT1-TSS1 0.00565028776 . . 1
chr8 130594026 130594707 . -1 -1 chr8:130594026-130594707::PVT1-TSS1 0.0005465308981 . . 0
What should we do? We propose to manually replace the gene name PVT1-TSS1 with PVT1, which, this time, is found in our reference gnid_gname:
awk '$2 ~ /(^PVT1$)/' /work2/project/fragencode/data/species.bk/homo_sapiens/hg19.gencv19/homo_sapiens.gnid.gnname.tsvENSG00000249859.3 PVT1
So we do:
# verify it's ok / 3 occurences of "ENSG00000249859.3" should be found awk 'BEGIN{FS="\t"; OLS="\t"} {if($12!=""){print $0} else{print $0"ENSG00000249859.3"}}' 3863.fulco.bedpe |awk 'BEGIN{FS="\t"} {occurence[$12]++} END{for(u in occurence){print u, occurence[u]}}' |sort -rnk2,2
If it's ok then:
mv 3863.fulco.bedpe temp && awk 'BEGIN{FS="\t"; OLS="\t"} {if($12!=""){print $0} else{print $0"ENSG00000249859.3"}}' temp > 3863.fulco.bedpe && rm temp
Now we need to sort it. In Fulco et al 2019 paper we found:
Genome build. All coordinates in the human genome are reported using build hg19.
So we use the following genome file: /work2/project/regenet/workspace/thoellinger/ABC-Enhancer-Gene-Prediction/reference/chr_sizes. Now:
srun --mem=8G --pty bash
conda activate base && module load bioinfo/bedtools-2.27.1
bedtools sort -faidx /work2/project/regenet/workspace/thoellinger/ABC-Enhancer-Gene-Prediction/reference/chr_sizes -i 3863.fulco.bedpe > 3863.fulco.bedpe.sorted
bedtools sort -faidx /work2/project/regenet/workspace/thoellinger/ABC-Enhancer-Gene-Prediction/reference/chr_sizes -i /work2/project/regenet/workspace/thoellinger/BENGI/Benchmark/Annotations/hg19-cCREs.bed > /work2/project/regenet/workspace/thoellinger/BENGI/Benchmark/Annotations/hg19-cCREs.sorted.bed
Finally:
bedtools intersect -sorted -wo -a 3863.fulco.bedpe.sorted -b /work2/project/regenet/workspace/thoellinger/BENGI/Benchmark/Annotations/hg19-cCREs.sorted.bed -g /work2/project/regenet/workspace/thoellinger/ABC-Enhancer-Gene-Prediction/reference/chr_sizes > 3863_ccRE_intersect.fulco.tsv
Remark: I don't really understand why it was necessary to provide the genome file for bedtools intersect (it did not work without the genome file). Until now, I think I never had to provide the genome file when using bedtools intersect.
$ wc -l 3863_ccRE.fulco.tsv 5111$ head -n 1 3863_ccRE_intersect.fulco.tsvchr1 3691278 3691778 . -1 -1 chr1:3691278-3691778::CEP104 0.02606174446 . . 15.67532312 8.056414 chr1 3690934 3693352 EH37D0003113 EH37E1056193 Promoter-like 500
Now, when there are more than 1 line for a single entry of Fulco et al. validation dataset, we keep only the one that maximize the intersect with the ccRE (note: later on we shall verify that it is okay to use all ccRE - I mean, if it would not be better to use only ccRE-dELS, to prevent reducing the intersection if we need to evaluate the predictions on the BENGI datasets...)
Doing so, only 3672 lines remain: we lost 191 pairs. Among these pairs, 103 positives remain.
awk 'BEGIN{FS="\t"; OFS="\t"} {if($19>intersection[$7]){intersection[$7]=$19; line[$7]=$17"\t"$12"\t"$11}} END{for(u in line){print line[u]}}' 3863_ccRE_intersect.fulco.tsv > 3672_ccRE_gene_pairs.fulco.txt
Partial reimplementation¶
Introduction¶
First of all we made a copy of all scripts, ie we duplicated Scripts folder in a new folder which we named local_Scripts.
Content of Run-Distance-Method.sh¶
We adapted of local_Scripts/Unsupervised-Methods/Run-Distance-Method.sh in local_Scripts/Unsupervised-Methods/CRISPRiFF_Run-Distance-Method.sh to use the CRISPRi-FlowFISH validation dataset.
#!/bin/bash
# Inserm computer
#workDir=~/Documents
# Personal computer
#workDir=~/Documents/INSERM
# Genotoul
workDir=/work2/project/regenet/workspace/thoellinger
setDir=$workDir/CRISPRi_FlowFISH
train=$setDir/3672_ccRE_gene_pairs.fulco.txt
outputDir=$workDir/BENGI/Distance-Method/Results
ccres=$workDir/BENGI/Benchmark/Annotations/hg19-cCREs.bed
scriptDir=$workDir/BENGI/local_Scripts/Unsupervised-Methods
tss=$workDir/BENGI/Benchmark/Annotations/GENCODEv19-TSSs.bed
mkdir -p $outputDir
python $scriptDir/rank.distance.py $tss $ccres \
$train $outputDir/K562.CRISPRi-FlowFISH-Distance.txt.unsorted
sort -t $'\t' -k 3,4 $outputDir/K562.CRISPRi-FlowFISH-Distance.txt.unsorted > $outputDir/K562.CRISPRi-FlowFISH-Distance.txt
rm $outputDir/K562.CRISPRi-FlowFISH-Distance.txt.unsorted
Content of rank.distance.py.sh¶
We replaced the content of rank.distance.py.sh with the following (the 2 columns we added in the output basically change nothing in the results, but are going to help us make sure results are sorted correctly when we are going to use them as an input for the Average Rank method. Still, that's not strictly necessary).
import sys, numpy as np
def Create_TSS_Dict(tss):
tss=open(tss)
tssDict={}
for line in tss:
line=line.rstrip().split("\t")
if line[6] in tssDict:
tssDict[line[6]].append(int(line[1]))
else:
tssDict[line[6]]=[int(line[1])]
tss.close()
return tssDict
def Create_Enhancer_Dict(enhancers):
enhancers=open(enhancers)
enhancerDict={}
for line in enhancers:
line=line.rstrip().split("\t")
enhancerDict[line[4]]=[int(line[1]),int(line[2])]
enhancers.close()
return enhancerDict
tss=sys.argv[1]
tssDict=Create_TSS_Dict(tss)
enhancers=sys.argv[2]
enhancerDict=Create_Enhancer_Dict(enhancers)
links=open(sys.argv[3])
output=open(sys.argv[4], "w+")
distanceArray=[]
for line in links:
line=line.rstrip().split("\t")
m=1000000000000
for x in tssDict[line[1].rstrip()]:
a=min([abs(enhancerDict[line[0].rstrip()][0]-x),abs(enhancerDict[line[0].rstrip()][1]-x)])
if a < m:
m=a
if m == 0:
print >> output, line[2]+"\t"+str(1)+"\t"+line[0]+"\t"+line[1]
else:
print >> output, line[2]+"\t"+str(1/float(m))+"\t"+line[0]+"\t"+line[1]
# if m == 0:
# print >> output, line[2]+"\t"+str(1)
# else:
# print >> output, line[2]+"\t"+str(1/float(m))
distanceArray.append(m)
links.close()
output.close()
Running the code¶
conda activate base && conda activate py2
./local_Scripts/Unsupervised-Methods/CRISPRiFF_Run-Distance-Method.sh
Analysis with R¶
Code (Rmd)¶
.badCode {
background-color: #C9DDE4;
}
library(knitr)
## Global options
options(max.print="75")
opts_chunk$set(echo=TRUE,
cache=FALSE,
prompt=FALSE,
tidy=TRUE,
comment=NA,
message=FALSE,
warning=FALSE,
class.source="badCode")
opts_knit$set(width=75)
library(ggplot2)
library(ggpubr) # for ggarrange
library(dplyr) # for bind_rows
# Tools for precision-recall : (see https://classeval.wordpress.com/tools-for-roc-and-precision-recall/)
library(precrec)
#library(ROCR)
#library(pROC)
#library(PRROC)
rm(list = ls())
# Personal
work_dir = "~/Documents/INSERM/"
# Inserm
#work_dir = "~/Documents/"
#####################
## Distance method ##
#####################
path_to_results = paste(work_dir, "BENGI/Distance-Method/Results/", sep='')
file_names = c("K562.CRISPRi-FlowFISH-Distance.txt")
short_names = c('CRiFF')
nb_files = length(file_names)
distance_colnames <- c('interaction', 'inverse.distance')
distance <- sapply(file_names, simplify=FALSE, function(file_name){
Df <- read.table(paste(path_to_results, as.character(file_name), sep=''), sep='\t')
Df[[1]] <- factor(Df[[1]], levels=c(0,1), labels=c("no interaction", "interaction"))
names(Df) <- distance_colnames
return(Df)
})
names(distance) <- short_names
#library(dplyr)
Distances <- bind_rows(distance, .id = 'method')
ggplot(aes(y = inverse.distance, x = method, fill = interaction), data = Distances) + geom_boxplot()
sscurves_distance <- list()
sscurves_distance <- sapply(distance, simplify=FALSE, function(Df){
evalmod(scores = Df$inverse.distance, labels = Df$interaction) # comes with "precrec" library
})
#library(ggplot2)
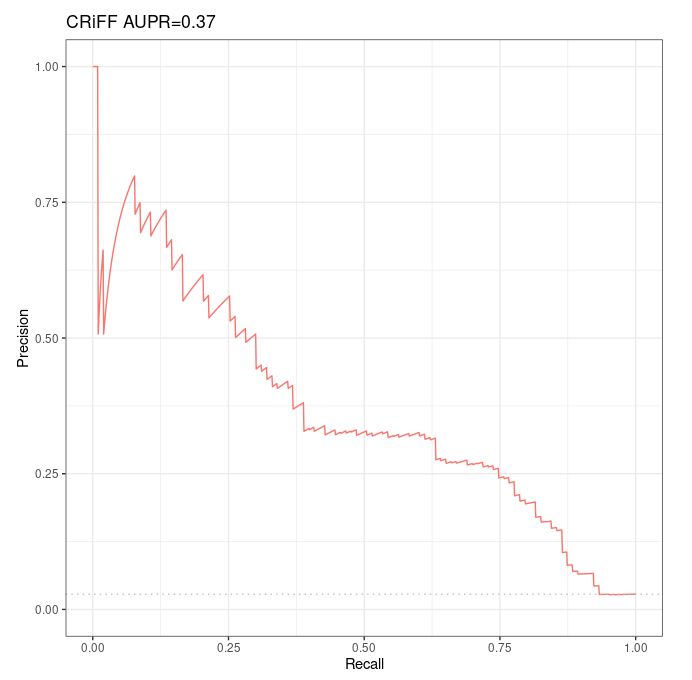
p1 <- autoplot(sscurves_distance[[1]], curvetype = c("PRC")) + ggtitle(paste(short_names[1], signif(attr(sscurves_distance[[1]], 'auc')[[4]][2], digits=2), sep = " AUPR="))
# ggarrange comes with library('ggpubr')
figure <- ggarrange(p1,
ncol = 1, nrow = 1)
figure
Results¶
Precision-Recall curves and AUPR of Moore et al's Distance method over Fulco et al validation dataset intersected with ccRE (103 positives and 3569 negatives).